


![]()
Synthetic Circuits and Regulatory Networks
Design of Synthetic Circuits
It has been widely acknowledged that the task of building circuits to meet multiple inducer-specific requirements is a challenging one. This is because of the incomplete description of component interactions compounded by the fact that the number of ways in which one can chose and interconnect components increases exponentially with the number of components. We have developed the OptCircuit framework that automatically identifies the circuit components and connectivity that brings about the desired functionality. Multiple literature sources are used to compile a comprehensive compilation of kinetic descriptions of promoter-protein pairs. The dynamics that govern the interactions between the elements of the genetic circuit are currently modeled using deterministic ordinary differential equations but the framework is general enough to accommodate stochastic simulations. The desired circuit response is abstracted as the maximization/minimization of an appropriately constructed objective function. Computational results for a toggle switch example demonstrate the ability of the framework to generate the complete list of circuit designs of varying complexity that exhibit the desired response. Designs identified for a genetic decoder highlight the ability of OptCircuit to suggest circuit configurations that go beyond the ones compatible with digital logic-based design principles. Finally, the results obtained from the concentration band detector example demonstrate the ability of OptCircuit to design circuits whose responses are contingent on the level of external inducer as well as pinpoint parameters for modification to rectify an existing (non-functional) biological circuit and restore functionality.
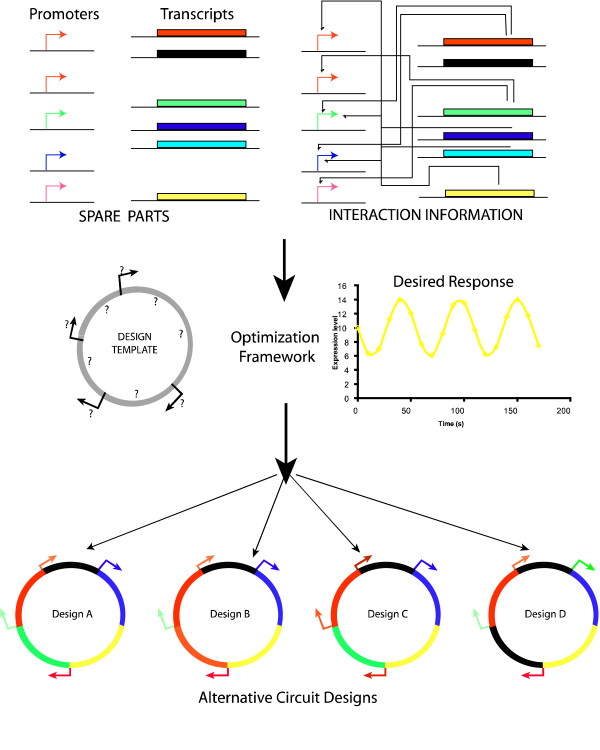
Figure Caption: A pictoral illustration of the OptCircuit framework. The three key components of the framework are the basic genetic elements (promoters, transcripts, inducers); the underlying kinetic mechanisms that drive the circuit response and finally the desired behavior of the circuit under construction. These three components are integrated by OptCircuit using an optimization based formulation.
[Dasika and Maranas (2008)]
Hierarchical methods for Regulatory Network Inference
It is now possible to generate genome-scale DNA microarray data for a variety of environmental and genetic perturbations. Microarray data, in conjunction with other sources of high throughput data (such as protein-protein interaction data and operon organization data), when coupled with sophisticated computational frameworks, promises to yield unprecedented insights into the organization and functioning of biological systems. However, in order to fully uncover the biological knowledge embedded in the experimental data, computational frameworks must be able to recognize and exploit the hierarchical nature of information flow and modularity in biological systems. In our group, we are pursuing the development of an integrated framework that will bring to bear both "top-down" and "bottom-up" approaches to generate candidate regulatory networks consistent with multiple types of data sets and subsequently test their mechanistic consistency (see also the following Figure). The overall goal is first to construct and then to apply an integrated computational infrastructure on genome-scale DNA microarray data available in the open literature and also provided by collaborating experimental labs to extend and subsequently assess the inference limits of modeling and computations.
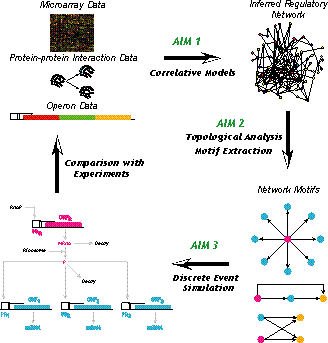
Figure Caption: Overview of research conducted in the area of gene regulatory network modeling
Of specific interest to us are the following research directions:
-
"Top-down" correlative inference of gene regulatory networks: We have developed linear and nonlinear correlative models allowing for time delay and employed them to elucidate putative regulatory networks that conform with multiple types of data such as DNA microarray, operon structure and protein-protein interactions. The microarray data used to this end is provided by Genencor International, Inc. The following figure highlights representative results for network topologies inferred for a selected set of 747 genes through the three different experimental data sets using the linear model are shown in the Figure below. The most striking feature of the inferred networks is the presence of "regulatory bands", a characteristic which indicates the existence of a small number of global regulators or "hubs" that influence a large number of other genes.
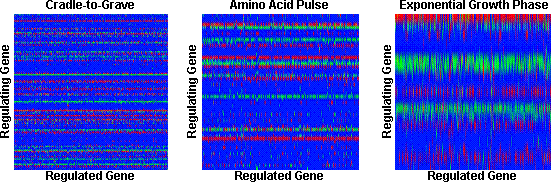
Figure Caption: Network topology inferred for the three experiments using the linear model. Green signifies activation or positive regulation whereas red denotes inhibition or negative regulation. Note that rows corresponding to genes not regulating any other genes are eliminated for clarity of presentation.
-
Analysis of topological properties of putative regulatory networks: Computational techniques are being customized and applied to the identification of the scaling characteristics of putative regulatory networks in collaboration with Dr. Reka Albert (Department of Physics, Penn State University). The goal of this part of our research is to decompose putative networks into clusters of motifs (sub-networks) for testing their mechanistic consistency through discrete event simulation procedures. The patterns to search for will range from simple two and three-node patterns, and to more complex ones as illustrated in the following Figure.

Figure Caption: Illustration of network motifs of increasing complexity. We look  for these motifs in the neighborhood of each gene and we will determine their appearance frequency.
-
"Bottom-up" mechanistic simulation of gene expression dynamics: We have developed a discrete event simulation platform DEMSIM for simulating small-to-medium scale gene regulatory networks. DEMSIM models the main processes in gene expression, which are transcription, translation and decay processes, as stand-alone modules while superimposing the regulatory circuitry to obtain an accurate time evolution of the system (see Figure below). The stochasticity inherent to gene expression and regulation processes is captured using Monte Carlo based sampling.The DEMSIM framework is implemented using the C programming language on a 16 node linux cluster. The proposed framework has been applied on a variety of regulatory network test systems.
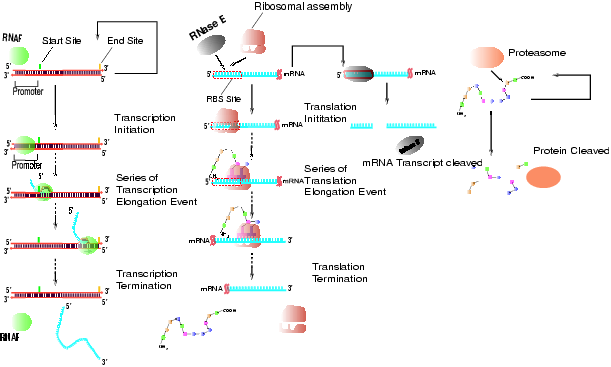
Figure Caption: Sequence of events governing (A) Transcription module (B) mRNA decay and Translation modules (C) Protein decay module
The overall objective of the research work is to derive to a new paradigm for a tightly integrated procedure for gene regulatory network elucidation where "top-down" model predictions are used to generate candidate networks consistent with the data and "bottom-up" detailed mechanistic simulations serve to verify, correct and complete the inferred networks.
[Gupta et al (2006); Christensen et al (2006); Gupta et al (2005); Dasika et al (2005); Dasika et al (2004)]