


![]()
Reconstruction, Analysis, & Redesign of Metabolic Pathways
Genome-scale Gene/Reaction Essentiality and Synthetic Lethality Analysis
Synthetic lethals refer to pairs of non-essential genes whose simultaneous deletion prohibits growth. One can extend the concept of synthetic lethality by considering gene groups of increasing size where only the simultaneous elimination of all genes is lethal whereas individual gene deletions are not. We developed optimization-based procedures for the exhaustive and targeted enumeration of multi-gene (and by extension multi-reaction) lethals for genome-scale metabolic models. Specifically, we are applying these approaches to the iAF1260 model of E. coli. This analysis for the iAF1260 model led to the complete identification of all double and triple synthetic lethals as well as the targeted identification of higher-order synthetic lethals. Graph representations of these synthetic lethals reveal a variety of motifs ranging from hub-like to highly connected sub-graphs providing a birds-eye view of the avenues available for redirecting metabolism and uncovering complex patterns of gene utilization and interdependence. The procedure also enables the use of falsely predicted synthetic lethals for metabolic model curation. By analyzing the functional classifications of the genes involved in synthetic lethals we reveal surprising connections within and across COG functional classifications.

Figure Caption: Topological and functional classification of clusters of SL gene pairs. Three types of network motifs are present: disjoint pairs, stars (1-connected motifs) and k-connected motifs (highly-connected subgraphs). Genes are color-coded in accordance to the COG (Tatusov et al, 2003) functional categorization.
Elucidation of Metabolic Fluxes using Labeled Isotopes
Atom mappings
A key consideration in metabolic engineering is the determination of fluxes of the metabolites within the cell. The gold standard in elucidating metabolic flows is the use of metabolic flux analysis (MFA). Flux elucidation requires both a detailed mapping of all possible paths different isotope forms can take through metabolic networks and experimental data in the form of labeled isotopes (usually 13C) sampled by NMR or GC-MS. When cells are fed a growth substrate with certain carbon positions labeled with 13C, the distribution of this label in the intracellular metabolites can be calculated based on the known biochemistry of the participating pathways. Most labeling studies have focused on skeletal representations of central metabolism that ignore many flux routes that could contribute to the observed isotopic labeling patterns. In contrast, our approach investigates the importance of carrying out isotopic labeling studies using a more complete reaction network. Our group has contributed and made available a large-scale isotope mapping model for E. coli spanning 238 reactions and 184 metabolites yielding 17,346 distinct isotopes by manually tracking the fate of labeled carbons. This network includes glycolysis, the TCA cycle, the pentose phosphate pathway, anaplerotic reactions, amino acid biosynthesis/ degradation, and oxidative phosphorylation. Many of these reactions are reversible and the model also includes a reaction to account for biomass production. Furthermore, the model includes global metabolite balances on cofactors such as ATP, NADH, and NADPH.

Figure Caption: The TCA cycle appears as a group of linked metabolites connected by reactions (lines) in a metabolic model (top). A carbon isotope model takes into account the additional linkages between the carbon atoms (numbered circles) and their transitions in the network (bottom). The symmetric nature of succinate (suc) and fumarate (fum) generates multiple path lines per atom. Some transitions are non-intuitive (e.g, accoa + oaa → cit), and all depend on the numbering scheme used for the atoms in each metabolite.
Flux elucidation
The experimental system was a strain of E. coli that produces amorphadiene, a precursor to the antimalarial drug artemisinin, grown on aerobic glucose conditions. We found that hundreds of distinct flux distributions were identified with sum of squared error (SSE) values that met the statistical cutoff for goodness-of-fit (i.e., chi-square). This lack of unique elucidation prompted us to deduce flux ranges spanned by equivalent solutions given the experimental error in the measurements. First, we calculated the variability in all fluxes subject to only network stoichiometry. As expected, nearly all of the central metabolic fluxes span wide ranges of values, motivating the need for 13C-based MFA. We subsequently recalculated flux variability after including the isotopomer balancing constraints to determine (i) which fluxes were fully resolved; (ii) how many of them were only partially resolved; and (iii) how many remaining fluxes in the model were completely unaffected by the isotopomer data.

Figure Caption: Flux map of only the central metabolism portion of the large-scale model. The net flux ranges are derived using optimization and incorporating the isotopic data. For reversible fluxes, a solid arrowhead indicates the forward direction.
Design of MFA experiments
One of the important considerations for analysis of the isotope model is how isotope measurements impact the elucidation of fluxes in large-scale metabolic reconstructions. This identifiability problem in MFA with isotopic considerations is very difficult, as isotopic balances yield nonlinear constraints. We have developed an integer programming framework, OptMeas, for the mathematical analysis of metabolic pathways to answer this question. By using a structural analysis-based optimization method, it is possible to exhaustively identify all combinations of isotope labeling experiments and flux measurement that completely resolve all flux values in the network. This approach results in an integer linear programming formulation while accounting for the case of partial measurements (e.g., when only some fragments are measured). These measurements, consisting of both partial or full isotope state determination, were assigned relative costs that allow the experimentalist to select the measurements that will be both sufficient and economical. The proposed framework has been tested on well-studied small demonstration examples. We have also benchmarked the proposed framework by applying it to medium-scale metabolic networks of E. coli and by revisiting our large-scale E. coli model to exhaustively identify all measurements options.

Figure Caption: OptMeas works by identifying the smallest measurement set that renders the incidence matrix of variables-equations full-column rank. By measuring fluxes and/or isotopomer distributions they cease to be variables implying that the corresponding columns in the structural matrix can be removed, eventually producing a full-column rank matrix that is structurally nonsingular.
[Chang et al (2008); Suthers et al (2007)]
Reconstruction of Genome-Scale Metabolic Models
There is growing interest in elucidating the minimal number of genes needed for life. This challenge is important not just for fundamental but also practical considerations arising from the need to design microorganisms exquisitely tuned for particular applications. With a genome size of ~580 kb and approximately 480 protein coding regions Mycoplasma genitalium is one of the smallest known self-replicating organisms and, additionally, has extremely fastidious nutrient requirements. The reduced genomic content of M. genitalium has led researchers to suggest that the molecular assembly contained in this organism may be a close approximation to the minimal set of genes required for bacterial growth. We have introduced a systematic approach for the construction and curation of a genome-scale in silico metabolic model for M. genitalium. The model accounts for 189 of the 482 genes listed in the latest genome annotation and is named iPS189. We used computation tools during the process to bridge network gaps in the model (i.e., GapFind and GapFill) and restore consistency with experimental data that determined which gene deletions led to cell death using GrowMatch. We achieved 87% correct model predictions for essential genes and 89% for non-essential genes. We subsequently have used the metabolic model to determine components that must be part of the growth medium. The general aproaches we used during the construction of the M. genitalium model are being used to develop models of other organisms.

Figure Caption: Classification of the ORFs included in iPS189 grouped into COG functional categories. The percent assigned to each class refers to the coverage of the total number in the genome accounted for in the model. Some of the ORFs in Mycoplasma genitalium do not currently have a COG functional category assignment (here represented as N/A). Note that although each ORF is only counted once within each COG functional category, some ORFs have multiple COG category assignments.
Over 700 genomes have been fully sequenced whereas only about 25 organism-specific genome-scale metabolic models have been constructed. The time required during the task of model-construction is becoming increasingly critical as genome sequencing for new organisms is proceeding at an ever-accelerating pace. The approaches and tools used during the M. genitalium model generation provide a roadmap for the automated metabolic reconstruction of other organisms. The application of the automated methodologies GapFind/GapFill and Growmatch can be effectively used during the construction process (as opposed to an a posteriori mode of deployment). Additionally, we have explored how GrowMatch is useful at improving model predictions when using growth phenotypes from various nutrient sources (i.e., carbon, nitrogen, phosphorus, sulfur) instead of single gene deletions.

Figure Caption: The four steps used during reconstruction are 1) identification of biotransformations using automated homology searches 2) assembly of reaction sets into a genome-scale metabolic model 3) automated network connectivity analysis and restoration, and 4) automated evaluation and improvement of model performance when compared to in vivo gene essentiality or growth data.
Curation of Genome-Scale Metabolic Models
Currently, there exists tens of different microbial and eukaryotic metabolic reconstructions (e.g.,Escherichia coli, Saccharomyces cerevisiae, Bacillus subtilis, Homo Sapiens) with many more under development. There are inaccuracies in these reconstructions due to the presence/absence of metabolic functionalities that the organism does not/ does actually possess. Our research is focused on developing optimization tools to pinpoint these inaccuracies and subsequently generate hypotheses to correct them. To this end, we have been involved in projects to develop the following tools:
1. GapFind/GapFill
Using GapFind , we identify the metabolites in the metabolic network reconstruction which cannot be produced under any uptake conditions. Subsequently, using GapFill, we identify the reactions from a customized multi-organism database that restores the connectivity of these metabolites to the parent network using four mechanisms. We demonstrate this procedure for the genome- scale reconstruction of Escherichia coli and also Saccharomyces cerevisiae wherein compartmentalization of intra-cellular reactions results in a more complex topology of the metabolic network. We determine that about 10% of metabolites in E. coli and 30% of metabolites in S. cerevisiae cannot carry any flux. Interestingly, the dominant flow restoration mechanism is directionality reversals of existing reactions in the respective models.

Figure Caption: GapFind identifies problem metabolites and GapFill generates hypotheses to restore the connectivity of these metabolites with the rest of the network.
2. GrowMatch
Genome-scale metabolic reconstructions are typically validated by comparing in silico growth predictions across different mutants utilizing different carbon sources with in vivo growth data. This comparison results in two types of model- prediction inconsistencies; either the model predicts growth when no growth is observed in the experiment (GNG inconsistencies) or the model predicts no growth when the experiment reveals growth (NGG inconsistencies). In this project, we developed an optimization-based framework, GrowMatch, to automatically reconcile GNG predictions (by suppressing functionalities in the model) and NGG predictions (by adding functionalities to the model). We use GrowMatch to resolve inconsistencies between the predictions of the latest in silico E. coli (iAF1260) model and the in vivo data available in the Keio collection and improved the consistency of in silico with in vivo predictions from 90.6% to 96.7%.
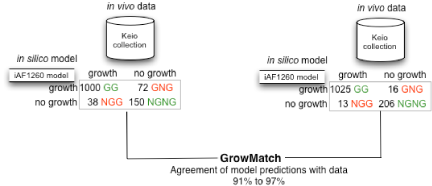
Figure Caption: Application of GrowMatch to the iAF1260 model to increase agreement of in silico predictions with in vivo results.
For many cases, GrowMatch identified fairly non-intuitive model modification hypotheses that would have been difficult to pinpoint through inspection alone. In addition, GrowMatch can be used during the construction phase of new, as opposed to existing, genome-scale metabolic models, leading to more expedient and accurate reconstructions.
[Satish Kumar and Maranas (2009); Satish Kumar et al (2007)]
Computational Procedures for Strain Optimization Using Stoichiometric Models of Metabolism
Our group introduced the computational framework termed OptKnock for suggesting gene deletion strategies that are likely to lead to the overproduction of specific chemical products. Specifically, a nested optimization framework (see Figure) was proposed to identify multiple gene deletion combinations that force the coupling of a cellular objective, here assumed to be a drain of biosynthetic precursors in the ratios required for biomass formation, with imposed chemical production targets. This coupling is accomplished by ensuring that the production of the desired product becomes an obligatory byproduct of growth by "shaping" the connectivity of the metabolic network. The computational procedure was designed to identify not just straightforward but also non-intuitive knockout strategies by considering the entire genome-scale metabolic networks as abstracted in recently developed in silico models.

Figure Caption: The bilevel optimization structure of OptKnock. The inner problem performs the flux allocation based on the optimization of a particular cellular objective (e.g., maximization of biomass yield, minimization of metabolic adjust, etc.). The outer problem then maximizes the bioengineering objective (e.g., compound overproduction) by restricting access to key reactions available to the optimization of the inner problem.
We have also developed the OptStrain framework which aims to guide pathways modifications, through reaction additions and deletions, of microbial networks for the overproduction of targeted compounds. These compounds may range from electrons or hydrogen in bio-fuel cell and environmental applications to complex drug precursor molecules. A comprehensive database of biotransformations, referred to as the Universal database (with over 5,000 reactions), is compiled and regularly updated by downloading and curating reactions from multiple biopathway database sources. Combinatorial optimization is then employed to elucidate the set(s) of non-native functionalities, extracted from this Universal database, to add to the examined production host for enabling the desired product formation. Subsequently, competing functionalities that divert flux away from the targeted product are identified and removed to ensure higher product yields coupled with growth. This work represents an advancement over earlier efforts by establishing an integrated computational framework capable of constructing stoichiometrically balanced pathways, imposing maximum product yield requirements, pinpointing the optimal substrate(s), and evaluating different microbial hosts.

Figure Caption: Pictorial representation of the OptStrain procedure. Step 1 involves the curation of database(s) of reactions to compile the Universal database, which comprises only elementally balanced reactions. Step 2 identifies a maximum-yield path enabling the desired biotransformation from a substrate (e.g., glucose, methanol, xylose) to product (e.g., hydrogen, vanillin) without any consideration for the origin of reactions. Note that the white arrows represent native reactions of the host and the yellow arrows denote non-native reactions. Step 3 minimizes the reliance on non-native reactions, and Step 4 incorporates the non-native functionalities into the microbial host's stoichiometric model and applies the OptKnock procedure to identify and eliminate reactions competing with the targeted product. The red x's pinpoint the deleted reactions.
[Pharkya and Maranas (2006); Pharkya and Maranas (2005); Fong et al (2005); Pharkya et al (2004); Pharkya et al (2003); Burgard et al (2003); Burgard and Maranas (2001)]
Analysis of Network Properties of Metabolic Models
In this research, we have introduced the Flux Coupling Finder (FCF) framework for elucidating the topological and flux connectivity features of genome-scale metabolic networks. The framework is demonstrated on genome-scale metabolic reconstructions of Helicobacter pylori, Escherichia coli, and Saccharomyces cerevisiae. The analysis allows one to determine if any two metabolic fluxes, v1 and v2, are (i) directionally coupled, if a non-zero flux for v1 implies a non-zero flux for v2 but not necessarily the reverse; (ii) partially coupled, if a non-zero flux for v1 implies a non-zero, though variable, flux for v2 and vice-versa; or (iii) fully coupled, if a non-zero flux for v1 implies not only a non-zero but also a fixed flux for v2 and vice-versa. Flux coupling analysis also enables the global identification of blocked reactions, which are all reactions incapable of carrying flux under a certain condition, equivalent knockouts, defined as the set of all possible reactions whose deletion forces the flux through a particular reaction to zero, and sets of affected reactions denoting all reactions whose fluxes are forced to zero if a particular reaction is deleted. The FCF approach thus provides a novel and versatile tool for aiding metabolic reconstructions and guiding genetic manipulations.
In analogy with the FCF method for fluxes we have developed an analogous method for metabolite pools. Specifically, conservation relationships for metabolite concentrations are important biophysical barriers, selected through evolution, to protect cellular organisms from stresses (e.g., osmotic) and provide global metabolic regulation.. The conservation relationships are linear combinations of metabolite concentrations that do not change over time and are solely determined by the organism's stoichiometry and uptake/secretion transport conditions. To this end, we have introduced an optimization-based framework to elucidate and analyze conservation relationships for metabolite concentrations in the context of genome-scale reaction networks. The framework is comprised of Metabolite Concentration Coupling Analysis (MCCA) and the Minimal Conserved Pool Identification (MCPI) procedure.

Figure Caption: Pictorial overview of coupled metabolite pools for E. coli.
[Nikolaev et al (2005); Burgard et al (2004); Burgard et al (2001)]
Analysis and Redesign for Kinetic Models of Metabolism
Moving from stoichiometric to kinetic models of metabolism, in this research, we proposed a general computational procedure to determine which genes/enzymes should be eliminated, repressed or overexpressed to maximize the flux through a product of interest for general kinetic models. The procedure relies on the generalized linearization of a kinetic description of the investigated metabolic system and the iterative application of mixed-integer linear (MILP) optimization implemented using the MATLAB environment. The proposed computational procedure is a general approach that can be applied to any metabolic system for which a kinetic description is provided. All MATLAB input files are made available as supplementary material.

Figure Caption: Hierarchical procedure for identifying optimal gene manipulation strategies. At each iteration of the outer loop q manipulations are enforced and qmax is the maximum number of manipulations. At each iteration of the inner loop a cut is added to exclude the current solution in the next iteration. zq is the objective function of interventions of q manipulations and z*q-1is the best objective function value of the best intervention strategy of q-1 manipulations.
[Vital-Lopez et al (2006); Nikolaev et al (2005); Petkov and Maranas (1997)]